
Активные формы кислорода
_%E2%80%93_some_examples_with_Lewis_structures.png)
Активные формы кислорода (АФК, реактивные формы кислорода, РФК, англ. Reactive oxygen species, ROS) — включают ионы кислорода, свободные радикалы и перекиси как неорганического, так и органического происхождения. Это, как правило, небольшие молекулы с исключительной реактивностью благодаря наличию неспаренного электрона на внешнем электронном уровне.
В живой клетке
АФК постоянно образуются в живой клетке как продукты нормального метаболизма кислорода. Активные формы кислорода образуются также под действием ионизирующего излучения. Некоторые АФК могут играть роль медиаторов важных внутриклеточных сигнальных путей. Однако повышенная продукция АФК приводит к оксидативному стрессу. Нормальные функции АФК включают индукцию иммунной системы и мобилизацию систем ионного транспорта. Например, клетки крови на месте повреждения начинают продуцировать АФК, что рекрутирует тромбоциты, необходимые для начала процесса заживления раны. АФК также запускают программируемую клеточную смерть (апоптоз).
Антиоксидантная защита
Около 98 % от всего потребляемого клеткой кислорода восстанавливается в митохондриях до воды в процессе окислительного фосфорилирования, но 2 % за счёт побочных реакций, в основном в начале или в середине дыхательной цепи митохондрий, в том числе реакций, катализируемых цитохром с-оксидазой, превращаются сначала в частично восстановленный супероксид кислорода, а затем в гидроксильный радикал OH•, также относящийся к АФК[1]. Помимо митохондрий к образованию АФК в результате реакции Фентона может приводить накапливающийся в клетках липофусцин из-за своей способности соединяться с некоторыми переходными металлами[2][3]. Защита клетки от АФК осуществляется несколькими антиоксидантными ферментами (супероксиддисмутаза, каталаза и пероксиредоксины) и низкомолекулярными антиоксидантами (витамин С, глутатион, мочевая кислота). Кроме этого, антиоксидантными свойствами обладают полифенолы (например, аналоги некоторых компонентов красного вина).
Повреждающее действие

Воздействие АФК на клеточный метаболизм научно доказано и подтверждается фактическими данными различных исследований[4]. Они не только участвуют в апоптозе (программируемой клеточной гибели), но и приводят к положительным результатам, например к индукции защитных[5][6] генов хозяина и мобилизации ионных транспортёров. Это говорит о том, что они контролируют клеточные функции. В частности тромбоциты, которые участвуют в заживлении ран и обеспечении гомеостаза, выпускают АФК, чтобы привлечь еще больше тромбоцитов к месту повреждения. Здесь же прослеживается связь с адаптивной иммунной системой за счет привлечения лейкоцитов.
Активные формы кислорода принимают участие в клеточных процессах при различных воспалительных реакциях, в том числе при заболеваниях сердечно-сосудистой системы. Кроме того, они несут ответственность за потерю слуха вследствие повреждения улитки громкими звуками, из-за ототоксичности таких препаратов, как цисплатин, и при врожденной глухоте у животных и людей. АФК выступают посредником в процессе апоптоза или программируемой клеточной гибели и ишемии. В качестве примеров здесь выступают инсульт и инфаркт.
В целом деструктивное воздействие активных форм кислорода на клетку чаще всего заключается в[7]:
- Повреждении ДНК или РНК
- Окислении полиненасыщенных жирных кислот в липидах (перекисное окисление липидов)
- Окислении аминокислот в белках
- Окисленной деактивации определенных ферментов за счет окисления кофакторов.
Реакция на патогены
Когда растение распознает чужеродный патоген, оно прежде всего начинает быстро производить супероксид или перекись водорода для укрепления клеточной стенки. Это препятствует распространению патогена в другие части растения. Вокруг патогена фактически формируется сетка, ограничивающая его передвижение и репродукцию.
В хозяине-млекопитающем АФК включают антимикробную защиту[8]. Удостовериться в значимости этой защиты можно на примере пациентов с гранулематозной хронической болезнью, которые страдают от недостаточного производства АФК. Они крайне восприимчивы к различным инфекциям, в том числе вызванным микроорганизмами Salmonella enterica, Staphylococcus aureus, Serratia marcescens и Aspergillus spp.
Исследования гомеостаза пищеварительного тракта дрозофилы фруктовой (Drosophila melanogaster) показывают, что производство АФК выступает в качестве ключевого фактора иммунного ответа в кишечнике насекомого. АФК выполняют бактерицидную функцию, повреждая ДНК, РНК и белки, а также выступают в роли сигнальных молекул, которые запускают механизмы регенерации эпителия[9]. Урацил, который выбрасывают микроорганизмы, инициирует производство и последующую активность двойной оксидазы (Duox) – фермента, производящего АФК в кишечнике. Активность Duox соотносится с концентрацией урацила в кишечнике, в базальных условиях ее подавляет протеинкиназа MkP3. Жесткий контроль за Duox препятствует чрезмерному производству АФК и облегчает процесс распознавания полезных и вредоносных микроорганизмов в кишечнике[10].
Способ, которым АФК защищают хозяина от вредоносных микроорганизмов, еще не до конца изучен. Вероятнее всего он заключается в повреждении ДНК микробов. Исследования сальмонеллы показывают, что для защиты от смертельных атак АТФ ей требовались механизмы репарации ДНК. Роль АФК в механизмах противовирусной защиты можно пронаблюдать через Rig-подобную хеликазу-1 и митохондриальный антивирусный сигнальный белок. Высокий уровень АФК усиливает сигналы через эти митохондриальные антивирусные рецепторы, чтобы активировать регуляторный фактор интерферона (IRF) 3, IRF7 и ядерный фактор каппа В (NF-κB) и нейтрализовать вирусы[11]. Клетки эпителия дыхательных путей подключают митохондриальные АФК в ответ на инфекцию гриппа. Активность АФК влечет за собой стимуляцию интерферона III типа и нейтрализацию вирусов, ограничивая возможность репликации[12]. АФК участвуют в защите организма хозяина от микобактерий, хотя, вероятно, не убивают их напрямую. Скорее АФК воздействуют на АФК-зависимые передачи сигналов, например, для производства цитокинов, аутофагии и формирования гранулёмы[13][14].
Активные формы кислорода также задействованы в активации, анергии и апоптозе т-лимфоцитов[15].
Окислительные повреждения
У аэробных форм жизни энергию, необходимую для осуществления биологический функций, производят митохондрии по электрон-транспортной цепи. Активные формы кислорода (АФК), потенциально способные навредить клеткам, производятся вместе с энергией. АФК могут повредить липиды, ДНК, РНК и белки, что в теории способствует физиологическим изменения в процессе старения.
АФК возникают, как нормальный продукт клеточного метаболизма. Среди них главный зачинщик окислительного повреждения – перекись водорода (H2O2), которая возникает из выделяемого митохондриями супероксида. Каталаза и супероксиддисмутаза смягчают разрушающее действие перекиси водорода и супероксида соответственно, превращая эти вещества в кислород и перекись водорода (которая позже превращается в воду) – то есть они производят безвредные молекулы. Однако эти превращения оказываются эффективными не на 100%, и остатки пероксидов остаются в клетках. Хотя АФК являются продуктом нормального функционирования клеток, их избыток приводит к пагубным последствиям[16].
Нарушение когнитивных функций
С возрастом память человека ухудшается, что очевидно при таких дегенеративных заболеваниях, как болезнь Альцгеймера, которые сопровождаются накоплением окислительных повреждений. Современные исследования показывают, что аккумуляция АФК снижает физические показатели организма, так как окислительные повреждения способствуют одряхлению. В частности, скапливающиеся окислительные повреждения приводят к когнитивной дисфункции, что подтвердило исследование на старых крысах, которым давали митохондриальные метаболиты и затем заставляли проходить когнитивные тесты. Судя по результатам, крысы лучше справлялись с задачами после метаболитов, и можно предположить, что метаболиты устраняли окислительные повреждения и улучшали функции митохондрий[17]. Аккумуляция окислительный повреждений может влиять на эффективность работы митохондрий и ускорять производство АФК[18]. Скапливающиеся окислительные повреждения и их роль в процессе старения зависят от типа поврежденной ткани. Дополнительные испытания помогли определить, что окислительные повреждения несут ответственность за ухудшение работы мозга в связи с возрастом. У старых когтистых песчанок выше концентрация окисленных белков, чем у молодых грызунов. Лечение старых и молодых мышей с использованием спиновых ловушек привело к уменьшению окисленных белков у старых песчанок, но не оказало никакого эффекта на молодых. Кроме того, старые песчанки лучше справлялись с когнитивными тестами во время лечения, но у них сократились функциональные возможности, когда лечение прекратилось, и уровень окисленных белков снова возрос. Опираясь на эти результаты ученые заключили, что окисление клеточных белков потенциально связано с функционированием мозга[19].
Причина старения
Свободнорадикальная теория старения утверждает, что окислительные повреждения, вызванные активными формами кислорода, провоцируют ухудшение функций, характерных для старения организма. Результаты исследований на беспозвоночных говорят о том, что у животных, генетически предрасположенных к нехватке определенных антиоксидантных ферментов (таких как СОД (супероксиддисмутаза)), сокращалась продолжительность жизни (что можно было предположить исходя из теории), тем не менее обратная манипуляция для повышения антиоксидантных ферментов не оказала устойчивого воздействия на продолжительность жизни (хотя некоторые исследования на дрозофилах подтверждают, что продолжительность жизни можно увеличить за счет сверхэкспресии марганец-СОД и ферментов, участвующих в биосинтезе глутатиона). Этой теории противоречит тот факт, что удаление митохондриальной СОД2 увеличивает продолжительность жизни у Caenorhabditis elegans[20].
С мышами происходит прохожая история. Удаление антиоксидантных ферментов, как правило, приводит к сокращению продолжительности жизни, хотя исследования сверхэкспресии (за некоторыми недавними исключениями) не привели к ее устойчивому увеличению[21]. Исследование преждевременного старения на крысах выявило у преждевременно состарившихся модельных животных повышенный окислительный стресс, снижение активности антиоксидантных ферментов и существенные повреждения ДНК в новой коре и гиппокампе по сравнению с нормально стареющей контрольной группой крыс[22]. Маркер повреждения ДНК 8-OHdG (8-Оксо-2′-дезоксигуанозин) – это продукт взаимодействия АФК с ДНК. Многочисленные исследования показали, что содержание 8-OHdG с возрастом повышается в различных органах млекопитающих[23].
Онкологические заболевания
АФК постоянно появляются в биологической системе для выполнения регуляторных функций и затем выводятся из нее[24]. При нормальных физиологических условиях клетки контролируют уровень АФК, удерживая баланс между генерацией и удалением АФК через очистительные системы. Но в условиях окислительного стресса чрезмерное скопление АФК разрушает клеточные белки, липиды и ДНК, из-за чего в клетке возникают необратимые очаговые поражения, способствующие канцерогенезу.
Раковые клетки испытывают более сильный АФК-стресс по сравнению со здоровыми клетками частично вследствие онкогенной стимуляции, повышенной метаболической активности и сбоя в работе митохондрий. АФК – это палка о двух концах. С одной стороны, они способствуют выживанию раковых клеток, так как прогрессия клеточного цикла под действием факторов роста и рецепторных тирозинкиназ (RTK) для активации нуждается в АФК[25]. Хроническое воспаление – главный медиатор онкологических заболеваний – тоже проходит под руководством АФК. С другой стороны, повышенная концентрация АФК подавляет рост опухолей через устойчивую активацию ингибитора клеточного цикла[26][27] и запуска программы клеточной смерти и физиологического старения за счет повреждения макромолекул. Большинство химиотерапевтических и радиотерапевтических агентов фактически убивают раковые клетки, усугубляя АФК-стресс[28][29]. Способность раковых клеток проводить различие между АФК-сигналами выживания и апоптоза обусловливается дозой, продолжительностью, типом и локализацией производства АФК. Для выживания раковым клеткам требуется умеренная концентрация АФК, а слишком высокий уровень приводит к их гибели.
Метаболическая адаптация опухолевой ткани уравновешивает потребность клетки в энергии равнозначной по степени важности потребностью в макромолекулярных составляющих и строгом контроле окислительно-восстановительного баланса. В результате значительно усиливается производство НАДФН как кофактора с восстанавливающей способностью во многих ферментативных реакциях макромолекулярного биосинтеза. В то же время это соединение спасает клетки от избытка АФК, возникающего из-за быстрой пролиферации. Клетки уравновешивают пагубное воздействие АФК производством антиоксидантов, таких как глутатион восстановленный (GSH) и тиоредоксин (TRX), которым для поддержания активности требуется восстанавливающая способность НАДФН[30].
Большинство факторов риска развития онкологии взаимодействуют с клетками посредством АФК. АФК активируют различные транскрипционные факторы, такие как универсальный фактор транскрипции активированных В-клеток (NF-κB), активаторный белок-1 (АР-1), фактор, индуцируемый гипоксией 1-альфа, преобразователь сигнала и активатор транскрипции 3 (STAT3), что приводит к экспрессии белков, контролирующих воспаление, клеточной трансформации, выживанию опухолевых клеток, пролиферации опухолевых клеток, инвазии, ангиогенезу и метастазам. Также АФК контролируют экспрессию различных генов-суппрессоров опухолевого роста, таких как р53, белок ретинобластомы (ген Rb), гомолог фосфатазы и тензина (PTEN)[31].
Канцерогенез
Связанное с АФК окисление ДНК – одна из основных причин мутаций, которые приводят к нескольким типам повреждений ДНК, в том числе модификации слабых (8-оксогуанин и формамидопиримидин) и сильных (циклопурин и этеноаддукты) оснований, АР-сайтам, нетипичным однонитевым разрывам, ДНК-аддуктам и внутри- и межмолекулярным сшивкам[32]. Исследователи подсчитали, что эндогенные АФК, которые образуются в результате нормального клеточного метаболизма, модифицируют в одной клетке примерно 20 000 оснований ДНК в день. 8-оксогуанин встречается чаще среди всех различных наблюдаемых окисленных азотистых оснований. Во время репликации ДНК-полимераза ошибочно соединяет 8-оксогуанин с аденином, что приводит к трансверсии G→T. Возникающая в результате геномная нестабильность напрямую способствует канцерогенезу. Клеточная трансформация приводит к развитию рака, а взаимодействие атипичной изоформы протеинкиназы C тип дзета (PKC-ζ) с p47phox контролирует производство АФК и трансформацию из стволовых клеток опухолей через аварийную программу «пузырчатого щита»[33][34].
Пролиферация клеток
Неконтролируемая пролиферация – характерный признак раковых клеток. Экзогенные и эндогенные АФК усиливают пролиферацию раковых клеток. Тот факт, что АФК содействуют росту опухолей, дополнительно подтверждают дальнейшие наблюдения: агенты, потенциально способные ингибировать образование АФК также могут ингибировать пролиферацию раковых клеток[31]. Хотя АФК способствуют пролиферации опухолевых клеток, значительное повышение концентрации АФК связывают со спадом пролиферации за счет остановки фазы G2/M клеточного цикла, повышенного фосфорилирования мутантного белка при атаксии телеангиэктазии (АТМ), киназы контрольной точки 1 (Chk1), Chk 2 и сокращению продолжительности цикла деления клетки 25 гомолог C (CDC25)[35].
Гибель клеток
Смерть раковой клетки может наступить тремя путями: апоптоз, некроз и аутофагия. Избыток АФК вызывает апоптоз через внешний и внутренний путь[36]. При внешнем пути апоптоза АФК генерируется Fas-лигандом в качестве предшествующего события активации Fas посредством фосфорилирования, что необходимо для последующего привлечения Fas-ассоциированного белка с доменом смерти и каспазой 8, а также для индукции апоптоза[31]. На внутреннем пути АФК способствуют высвобождению цитохрома С за счет активации белков, стабилизирующих поры (Bcl-2 и Bcl-xL), а также блокирования белков, дестабилизирующих поры (Bcl-2-связанный Х белок, гомологичный белок-антагонист-киллер Bcl-2)[37]. Внутренний путь, который также называют каспазный каскад, начинается с повреждения митохондрий, которое запускает высвобождение цитохрома С. Повреждение ДНК, окислительный стресс и потеря потенциала мембраны митохондрий приводят к выделению вышеупомянутых проапоптотических белков, которые стимулируют апоптоз[38]. Повреждение митохондрий тесно связано с апоптозом, а поскольку митохондрии легко поразить, здесь существует потенциал для онкотерапии[39]. Цитотоксические свойства АФК выступают в качестве движущей силы апоптоза, но при еще более высокой концентрации АФК могут привести как к апоптозу, так и некрозу – неконтролируемой формы клеточной гибели – раковых клеток[40].
Многочисленные исследования указывают на пути и ассоциации уровня АФК с апоптозом, но новое направление исследований связывает концентрацию АФК и аутофагию[41]. АФК могут вызывать гибель клеток посредством аутофагии – самостоятельный катаболический процесс, который заключается в секвестрации цитоплазматического содержимого (истощенных или повреждённых органелл и агрегированных белков) и его деградации лизосомами[42]. Следовательно, аутофагия поддерживает здоровье клетки во время окислительного стресса. Уровни концентрации АФК могут вызвать аутофагию в клетке различными путями, чтобы избавиться от вредоносных органелл, например канцерогенов, и предотвратить повреждение, не вызывая апоптоза[43]. Аутофагическая гибель клеток может наступить в результате чрезмерной экспрессии аутофагии, когда клетка поглощает слишком много частей самой себя в попытке компенсировать повреждения и больше не способна продолжать жизнь. При этом типе гибели наблюдается усиление или потеря контроля над генами, регулирующими аутофагию[44]. Таким образом, как только исследователи глубже изучат процесс аутофагической гибели клеток и ее связи с АФК, эта форма программируемой клеточной гибели сможет послужить в качестве новой онкотерапии. Аутофагия и апоптоз – разные механизмы клеточной смерти, которые вызывает высокое содержание АФК. Однако аутофагия и апоптоз редко развиваются строго независимыми путями. Существует доказанная связь между АФК и аутофагией и корреляция между избыточным количеством АФК и апоптозом[43]. Деполяризация мембраны митохондрий – еще одна характеристика начала аутофагии. Когда поврежденные митохондрии начинают выделять АФК, инициируется аутофагия, чтобы избавиться от бесполезных органелл. Если препарат нацелен на митохондрии и создает АФК, то аутофагия поможет избавить клетку от большого количества митохондрий и других поврежденных органелл, которые оказались нежизнеспособными. Чрезмерное количество АФК и повреждение митохондрий могут также служить сигналом апоптоза. Баланс аутофагии внутри клетки и взаимовлияние аутофагии и апоптоза, опосредованное АФК, имеет решающее значение для выживания клетки. Взаимовлияние и связь между аутофагией и апоптозом могут стать мишенью для онкотерапий или найти применение в методах комбинированного лечения устойчивых видов рака.
Инвазия, ангиогенез и метастазы
После стимуляции фактора роста рецепторных тирозинкиназ (RTK), АФК могут запустить активацию сигнальных путей, участвующих в миграции и инвазии клеток, например из семейства митоген-активируемой протеинкиназы (MAPK) – киназа, регулируемая внеклеточными сигналами (ERK), c-jun NH-2-терминальная киназа и p38 MAPK. АФК способствуют миграции за счет усиления фосфорилирования киназы очаговой адгезии (FAK) p130Cas и паксилина[45].
В ходе исследований in vitro и in vivo выяснилось, что АФК индуцируют факторы транскрипции и модулируют сигнальные молекулы, участвующие в ангиогенезе (MMP, VEGF) и метастазировании (повышение экспрессии АР-1, CXCR4, AKT и подавление PTEN)[31].
Хроническое воспаление и рак
Экспериментальные и эпидемиологические исследования в течение последних нескольких лет позволили обнаружить близкие ассоциации между АФК, хроническим воспалением и раком[31]. АФК провоцируют хроническое воспаление через индукцию ЦОГ-2, воспалительных цитокинов (TNFα, интерлейкин 1 ((IL-1), IL-6, хемокинов (IL-8, CXCR4) и провоспалительных факторов транскрипции (NF-κB)[31]. Хемокины и хемокиновые рецепторы в свою очередь способствуют инвазии и метастазированию различных типов опухолей.
Лечение рака
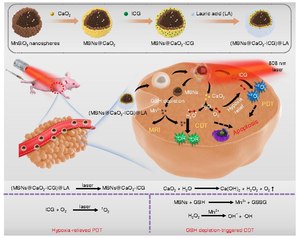
Существуют стратегии повышения и снижения уровней АФК, но чаще отдают предпочтения второй из них. Раковые клетки с высокой концентрацией АФК находятся в зависимости от антиоксидантной защитной системы. Препараты, повышающие уровень АФК, усиливают окислительный стресс либо путем прямого образования АФК (например мотексафин гадолиний и элескломол), либо с помощью агентов, которые отключают внутреннюю антиоксидантную систему, например ингибитор СОД (ATN-224, 2-метоксиэстрадиол) и ингибитор GSH (PEITC, бутионина сульфоксимин). В результате повышается уровень эндогенных АФК, которые при превышении порога переносимости могут вызвать гибель клетки[46]. С другой стороны, нормальные клетки при низком уровне стресса лучше справляются с дополнительными воздействиями, которые генерируют АФК, чем раковые клетки[47]. Таким образом повышение концентрации АФК во всех клетках можно рассматривать, как способ селективного уничтожения раковых клеток.
В основании лучевой терапии тоже лежит токсичность АФК, которая уничтожает опухолевые клетки. В лучевой терапии применяют рентгеновские лучи, гамма-лучи, а также излучения тяжелых частиц, таких как протоны и нейтроны, для индукции гибели клеток и митотической катастрофы, опосредованной АФК[31].
По причине двойной роли АФК были разработаны как прооксидантные, так и антиоксидантные противораковые средства. Однако сама по себе модуляция сигналов АФК не может считаться идеальным методом из-за адаптации раковых клеток к АФК-стрессу, избыточных путей поддержания роста опухоли и токсичности противораковых препаратов, генерирующих АФК. Необходимо комбинировать лекарства, генерирующие АФК, с фармацевтическими препаратами, которые могут нарушить окислительно-восстановительную адаптацию, чтобы выстроить наиболее удачную стратегию повышения цитотоксичности раковых клеток[31].
Джеймс Уотсон[48] совместно с коллегами[49] выдвинул предположение, что недостаток внутриклеточных АФК из-за отсутствия физических упражнений способствует малигнизации опухолей, поскольку для правильного фолдинга белка в эндоплазматическом ретикулуме необходима пиковая концентрация АФК. А низкий уровень АФК специфически препятствует образованию белков-супрессоров опухоли[49]. Поскольку физические упражнения вызывают временные всплески АФК, это объясняет тот факт, что физическая активность улучшает прогноз для онкологических больных.[50] Более того, препараты, которые сильно повышают уровень АФК (например, 2-дезокси-D-глюкоза и индукторы клеточного стресса на основе углеводов) эффективнее вызывают гибель раковых клеток из-за падкости раковых клеток на сахар[51].
См. также
Литература
- Free Radicals in Biology and Medicine by B.Halliwell and M.C.Gutteridge. Oxford University Press, 2000
- Sen, C.K. (2003) The general case for redox control of wound repair, Wound Repair and Regeneration, 11, 431—438
- Krötz, F., Sohn, HY., Gloe, T., Zahler, S., Riexinger, T., Schiele, T.M., Becker, B.F., Theisen, K., Klauss, V., Pohl, U. (2002) NAD(P)H oxidase-dependent platelet superoxide anion release increases platelet recruitment, Blood, 100, 917—924
- Pignatelli, P. Pulcinelli, F.M., Lenti, L., Gazzaniga, P.P., Violi, F. (1998) Hydrogen Peroxide Is Involved in Collagen-Induced Platelet Activation, Blood, 91 (2), 484—490
- Guzik, T.J., Korbut, R., Adamek-Guzik, T. (2003) Nitric oxide and superoxide in inflammation and immune regulation, Journal of Physiology and Pharmacology, 54 (4), 469—487
Примечания
- ↑ Скулачёв В. П. Эволюция, митохондрии кислород // Соросовский образовательный журнал : журнал. — 1999. — № 9. Архивировано 30 августа 2018 года.
- ↑ Annika Höhn, Tobias Jung, Stefanie Grimm, Tilman Grune. Lipofuscin-bound iron is a major intracellular source of oxidants: role in senescent cells // Free Radical Biology & Medicine. — 2010-04-15. — Т. 48, вып. 8. — С. 1100–1108. — ISSN 1873-4596. — doi:10.1016/j.freeradbiomed.2010.01.030. Архивировано 1 сентября 2018 года.
- ↑ Jeannette König, Christiane Ott, Martín Hugo, Tobias Jung, Anne-Laure Bulteau. Mitochondrial contribution to lipofuscin formation // Redox Biology. — 2017-01-25. — Т. 11. — С. 673–681. — ISSN 2213-2317. — doi:10.1016/j.redox.2017.01.017.
- ↑ Kannan Muthukumar, Vasanthi Nachiappan. Cadmium-induced oxidative stress in Saccharomyces cerevisiae // Indian Journal of Biochemistry & Biophysics. — 2010-12. — Т. 47, вып. 6. — С. 383–387. — ISSN 0301-1208.
- ↑ Balázs Rada, Thomas L. Leto. Oxidative innate immune defenses by Nox/Duox family NADPH oxidases // Contributions to Microbiology. — 2008. — Т. 15. — С. 164–187. — ISSN 1420-9519. — doi:10.1159/000136357.
- ↑ Gregory E. Conner, Matthias Salathe, Rosanna Forteza. Lactoperoxidase and hydrogen peroxide metabolism in the airway // American Journal of Respiratory and Critical Care Medicine. — 2002-12-15. — Т. 166, вып. 12 Pt 2. — С. S57–61. — ISSN 1073-449X. — doi:10.1164/rccm.2206018.
- ↑ Robert J. Brooker. Genetics : analysis & principles. — 4th ed. — New York: McGraw-Hill, 2012. — xviii, 761, [85] pages с. — ISBN 0-07-352528-6, 978-0-07-352528-0.
- ↑ Marc Herb, Michael Schramm. Functions of ROS in Macrophages and Antimicrobial Immunity // Antioxidants (Basel, Switzerland). — 2021-02-19. — Т. 10, вып. 2. — С. 313. — ISSN 2076-3921. — doi:10.3390/antiox10020313.
- ↑ Nicolas Buchon, Nichole A. Broderick, Bruno Lemaitre. Gut homeostasis in a microbial world: insights from Drosophila melanogaster // Nature Reviews. Microbiology. — 2013-09. — Т. 11, вып. 9. — С. 615–626. — ISSN 1740-1534. — doi:10.1038/nrmicro3074.
- ↑ Kyung-Ah Lee, Sung-Hee Kim, Eun-Kyoung Kim, Eun-Mi Ha, Hyejin You. Bacterial-derived uracil as a modulator of mucosal immunity and gut-microbe homeostasis in Drosophila // Cell. — 2013-05-09. — Т. 153, вып. 4. — С. 797–811. — ISSN 1097-4172. — doi:10.1016/j.cell.2013.04.009.
- ↑ West AP et al 2011 Nature Reviews Immunology 11. — С. 389–402.
- ↑ Hyun Jik Kim, Chang-Hoon Kim, Ji-Hwan Ryu, Min-Ji Kim, Chong Yoon Park. Reactive oxygen species induce antiviral innate immune response through IFN-λ regulation in human nasal epithelial cells // American Journal of Respiratory Cell and Molecular Biology. — 2013-11. — Т. 49, вып. 5. — С. 855–865. — ISSN 1535-4989. — doi:10.1165/rcmb.2013-0003OC.
- ↑ Marc Herb, Alexander Gluschko, Katja Wiegmann, Alina Farid, Anne Wolf. Mitochondrial reactive oxygen species enable proinflammatory signaling through disulfide linkage of NEMO // Science Signaling. — 2019-02-12. — Т. 12, вып. 568. — С. eaar5926. — ISSN 1937-9145. — doi:10.1126/scisignal.aar5926.
- ↑ Christine Deffert, Julien Cachat, Karl-Heinz Krause. Phagocyte NADPH oxidase, chronic granulomatous disease and mycobacterial infections // Cellular Microbiology. — 2014-08. — Т. 16, вып. 8. — С. 1168–1178. — ISSN 1462-5822. — doi:10.1111/cmi.12322.
- ↑ Aleksey V. Belikov, Burkhart Schraven, Luca Simeoni. T cells and reactive oxygen species // Journal of Biomedical Science. — 2015-10-15. — Т. 22. — С. 85. — ISSN 1423-0127. — doi:10.1186/s12929-015-0194-3.
- ↑ Understanding the process of aging : the roles of mitochondria, free radicals, and antioxidants. — New York: Marcel Dekker, 1999. — 1 online resource (xv, 366 pages) с. — ISBN 0-585-13410-3, 978-0-585-13410-9.
- ↑ Jiankang Liu, Elizabeth Head, Afshin M. Gharib, Wenjun Yuan, Russell T. Ingersoll. Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: partial reversal by feeding acetyl-L-carnitine and/or R-alpha -lipoic acid // Proceedings of the National Academy of Sciences of the United States of America. — 2002-02-19. — Т. 99, вып. 4. — С. 2356–2361. — ISSN 0027-8424. — doi:10.1073/pnas.261709299.
- ↑ Earl R. Stadtman. Protein Oxidation and Aging (англ.) // Science. — 1992-08-28. — Vol. 257, iss. 5074. — P. 1220–1224. — ISSN 1095-9203 0036-8075, 1095-9203. — doi:10.1126/science.1355616.
- ↑ J. M. Carney, P. E. Starke-Reed, C. N. Oliver, R. W. Landum, M. S. Cheng. Reversal of age-related increase in brain protein oxidation, decrease in enzyme activity, and loss in temporal and spatial memory by chronic administration of the spin-trapping compound N-tert-butyl-alpha-phenylnitrone // Proceedings of the National Academy of Sciences of the United States of America. — 1991-05-01. — Т. 88, вып. 9. — С. 3633–3636. — ISSN 0027-8424. — doi:10.1073/pnas.88.9.3633.
- ↑ Jeremy M. Van Raamsdonk, Siegfried Hekimi. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans // PLoS genetics. — 2009-02. — Т. 5, вып. 2. — С. e1000361. — ISSN 1553-7404. — doi:10.1371/journal.pgen.1000361.
- ↑ Florian L. Muller, Michael S. Lustgarten, Youngmok Jang, Arlan Richardson, Holly Van Remmen. Trends in oxidative aging theories // Free Radical Biology & Medicine. — 2007-08-15. — Т. 43, вып. 4. — С. 477–503. — ISSN 0891-5849. — doi:10.1016/j.freeradbiomed.2007.03.034.
- ↑ J. K. Sinha, S. Ghosh, U. Swain, N. V. Giridharan, M. Raghunath. Increased macromolecular damage due to oxidative stress in the neocortex and hippocampus of WNIN/Ob, a novel rat model of premature aging // Neuroscience. — 2014-06-06. — Т. 269. — С. 256–264. — ISSN 1873-7544. — doi:10.1016/j.neuroscience.2014.03.040.
- ↑ New research on DNA damage. — New York: Nova Science Publishers, 2008. — xvi, 410 pages с. — ISBN 978-1-60456-581-2, 1-60456-581-0.
- ↑ Bryan C. Dickinson, Christopher J. Chang. Chemistry and biology of reactive oxygen species in signaling or stress responses // Nature Chemical Biology. — 2011-07-18. — Т. 7, вып. 8. — С. 504–511. — ISSN 1552-4469. — doi:10.1038/nchembio.607.
- ↑ K. Irani, Y. Xia, J. L. Zweier, S. J. Sollott, C. J. Der. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts // Science (New York, N.Y.). — 1997-03-14. — Т. 275, вып. 5306. — С. 1649–1652. — ISSN 0036-8075. — doi:10.1126/science.275.5306.1649.
- ↑ Matthew R. Ramsey, Norman E. Sharpless. ROS as a tumour suppressor? // Nature Cell Biology. — 2006-11. — Т. 8, вып. 11. — С. 1213–1215. — ISSN 1465-7392. — doi:10.1038/ncb1106-1213.
- ↑ Akiko Takahashi, Naoko Ohtani, Kimi Yamakoshi, Shin-ichi Iida, Hidetoshi Tahara. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence // Nature Cell Biology. — 2006-11. — Т. 8, вып. 11. — С. 1291–1297. — ISSN 1465-7392. — doi:10.1038/ncb1491.
- ↑ Markus F. Renschler. The emerging role of reactive oxygen species in cancer therapy // European Journal of Cancer (Oxford, England: 1990). — 2004-09. — Т. 40, вып. 13. — С. 1934–1940. — ISSN 0959-8049. — doi:10.1016/j.ejca.2004.02.031.
- ↑ Steven M. Toler, Dennis Noe, Amarnath Sharma. Selective enhancement of cellular oxidative stress by chloroquine: implications for the treatment of glioblastoma multiforme // Neurosurgical Focus. — 2006-12-15. — Т. 21, вып. 6. — С. E10. — ISSN 1092-0684. — doi:10.3171/foc.2006.21.6.1.
- ↑ Rob A. Cairns, Isaac S. Harris, Tak W. Mak. Regulation of cancer cell metabolism // Nature Reviews. Cancer. — 2011-02. — Т. 11, вып. 2. — С. 85–95. — ISSN 1474-1768. — doi:10.1038/nrc2981.
- ↑ 31,0 31,1 31,2 31,3 31,4 31,5 31,6 31,7 Subash C. Gupta, David Hevia, Sridevi Patchva, Byoungduck Park, Wonil Koh. Upsides and downsides of reactive oxygen species for cancer: the roles of reactive oxygen species in tumorigenesis, prevention, and therapy // Antioxidants & Redox Signaling. — 2012-06-01. — Т. 16, вып. 11. — С. 1295–1322. — ISSN 1557-7716. — doi:10.1089/ars.2011.4414.
- ↑ Gulam Waris, Haseeb Ahsan. Reactive oxygen species: role in the development of cancer and various chronic conditions // Journal of Carcinogenesis. — 2006-05-11. — Т. 5. — С. 14. — ISSN 1477-3163. — doi:10.1186/1477-3163-5-14.
- ↑ Goodwin G. Jinesh, Rikiya Taoka, Qiang Zhang, Siddharth Gorantla, Ashish M. Kamat. Novel PKC-ζ to p47 phox interaction is necessary for transformation from blebbishields // Scientific Reports. — 2016-04-04. — Т. 6. — С. 23965. — ISSN 2045-2322. — doi:10.1038/srep23965.
- ↑ G. G. Jinesh, A. M. Kamat. Blebbishield emergency program: an apoptotic route to cellular transformation // Cell Death and Differentiation. — 2016-05. — Т. 23, вып. 5. — С. 757–758. — ISSN 1476-5403. — doi:10.1038/cdd.2016.26.
- ↑ B. N. Ames. Dietary carcinogens and anticarcinogens. Oxygen radicals and degenerative diseases // Science (New York, N.Y.). — 1983-09-23. — Т. 221, вып. 4617. — С. 1256–1264. — ISSN 0036-8075. — doi:10.1126/science.6351251.
- ↑ Tomris Ozben. Oxidative stress and apoptosis: impact on cancer therapy // Journal of Pharmaceutical Sciences. — 2007-09. — Т. 96, вып. 9. — С. 2181–2196. — ISSN 0022-3549. — doi:10.1002/jps.20874.
- ↑ Jennifer L. Martindale, Nikki J. Holbrook. Cellular response to oxidative stress: signaling for suicide and survival // Journal of Cellular Physiology. — 2002-07. — Т. 192, вып. 1. — С. 1–15. — ISSN 0021-9541. — doi:10.1002/jcp.10119.
- ↑ M. Chiara Maiuri, Einat Zalckvar, Adi Kimchi, Guido Kroemer. Self-eating and self-killing: crosstalk between autophagy and apoptosis // Nature Reviews. Molecular Cell Biology. — 2007-09. — Т. 8, вып. 9. — С. 741–752. — ISSN 1471-0080. — doi:10.1038/nrm2239.
- ↑ Simone Fulda, Lorenzo Galluzzi, Guido Kroemer. Targeting mitochondria for cancer therapy // Nature Reviews. Drug Discovery. — 2010-06. — Т. 9, вып. 6. — С. 447–464. — ISSN 1474-1784. — doi:10.1038/nrd3137.
- ↑ M. B. Hampton, S. Orrenius. Dual regulation of caspase activity by hydrogen peroxide: implications for apoptosis // FEBS letters. — 1997-09-15. — Т. 414, вып. 3. — С. 552–556. — ISSN 0014-5793. — doi:10.1016/s0014-5793(97)01068-5.
- ↑ Spencer B. Gibson. A matter of balance between life and death: targeting reactive oxygen species (ROS)-induced autophagy for cancer therapy // Autophagy. — 2010-10. — Т. 6, вып. 7. — С. 835–837. — ISSN 1554-8635. — doi:10.4161/auto.6.7.13335.
- ↑ Ashutosh Shrivastava, Paula M. Kuzontkoski, Jerome E. Groopman, Anil Prasad. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy // Molecular Cancer Therapeutics. — 2011-07. — Т. 10, вып. 7. — С. 1161–1172. — ISSN 1538-8514. — doi:10.1158/1535-7163.MCT-10-1100.
- ↑ 43,0 43,1 Ruth Scherz-Shouval, Zvulun Elazar. ROS, mitochondria and the regulation of autophagy // Trends in Cell Biology. — 2007-09. — Т. 17, вып. 9. — С. 422–427. — ISSN 1879-3088. — doi:10.1016/j.tcb.2007.07.009.
- ↑ Zhiping Xie, Daniel J. Klionsky. Autophagosome formation: core machinery and adaptations // Nature Cell Biology. — 2007-10. — Т. 9, вып. 10. — С. 1102–1109. — ISSN 1465-7392. — doi:10.1038/ncb1007-1102.
- ↑ Lalchhandami Tochhawng, Shuo Deng, Shazib Pervaiz, Celestial T. Yap. Redox regulation of cancer cell migration and invasion // Mitochondrion. — 2013-05. — Т. 13, вып. 3. — С. 246–253. — ISSN 1872-8278. — doi:10.1016/j.mito.2012.08.002.
- ↑ Paul T. Schumacker. Reactive oxygen species in cancer cells: live by the sword, die by the sword // Cancer Cell. — 2006-09. — Т. 10, вып. 3. — С. 175–176. — ISSN 1535-6108. — doi:10.1016/j.ccr.2006.08.015.
- ↑ Dunyaporn Trachootham, Jerome Alexandre, Peng Huang. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? // Nature Reviews. Drug Discovery. — 2009-07. — Т. 8, вып. 7. — С. 579–591. — ISSN 1474-1784. — doi:10.1038/nrd2803.
- ↑ James D. Watson. Type 2 diabetes as a redox disease // Lancet (London, England). — 2014-03-01. — Т. 383, вып. 9919. — С. 841–843. — ISSN 1474-547X. — doi:10.1016/S0140-6736(13)62365-X.
- ↑ 49,0 49,1 Remco J. Molenaar, Cornelis J. van Noorden. Type 2 diabetes and cancer as redox diseases? // Lancet (London, England). — 2014-09-06. — Т. 384, вып. 9946. — С. 853. — ISSN 1474-547X. — doi:10.1016/S0140-6736(14)61485-9.
- ↑ Melinda L. Irwin, Ashley Wilder Smith, Anne McTiernan, Rachel Ballard-Barbash, Kathy Cronin. Influence of pre- and postdiagnosis physical activity on mortality in breast cancer survivors: the health, eating, activity, and lifestyle study // Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology. — 2008-08-20. — Т. 26, вып. 24. — С. 3958–3964. — ISSN 1527-7755. — doi:10.1200/JCO.2007.15.9822.
- ↑ Fidelis T. Ndombera, Garrett C. VanHecke, Shima Nagi, Young-Hoon Ahn. Carbohydrate-based inducers of cellular stress for targeting cancer cells // Bioorganic & Medicinal Chemistry Letters. — 2016-03-01. — Т. 26, вып. 5. — С. 1452–1456. — ISSN 1464-3405. — doi:10.1016/j.bmcl.2016.01.063.